Biologists’ Mapping Method Illustrates Paths to New Proteins
By: Stephen Fontenot | July 7, 2023

Scientists at The University of Texas at Dallas are using machine learning to study proteins — the molecules that carry out essential life functions — in a way that could impact protein engineering, human health and the evolutionary tracking of proteins related to infectious diseases.
In the growing field of protein design, researchers examine the evolutionary history of proteins — how their structure and function have changed over time due to genetic mutations — and could use that information to potentially design new proteins for purposes like fighting diseases or enabling biotechnology applications from novel proteins not existent in nature.
A team led by Dr. Faruck Morcos, associate professor of biological sciences in the School of Natural Sciences and Mathematics, is using advanced computer techniques to generate a 3D “landscape” that allows scientists to visualize how viable new proteins could be engineered.

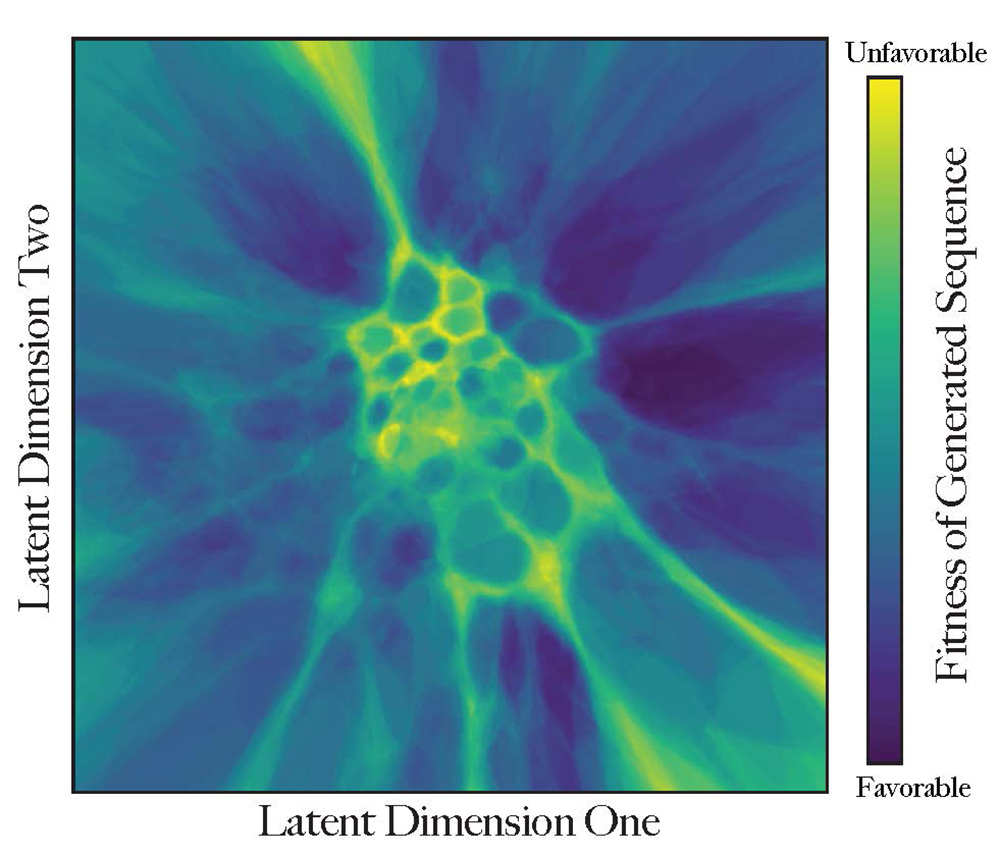
“This latent generative landscape represents an advancement in the modeling of proteins and, together with the software we have published, is an accessible tool for those seeking to generate, engineer or study proteins and their functions,” said computational biology doctoral student Cheyenne Ziegler MS’20, one of the lead authors of a paper published online April 19 in Nature Communications describing the work. Morcos is the corresponding author of the study.
Proteins are made up of sequences of molecular building blocks called amino acids. Protein sequences give researchers clues to their functions in the body.
“Our new framework is like a road map,” Morcos said. “Rather than simply analyzing existing protein sequences, we look at the evolution of the proteins and construct maps looking both at proteins that already exist as well as generating and plotting out potential sequences.”
Using variational autoencoders (VAE) — an unsupervised learning model incorporating a neural network and coevolutionary modeling, an inference technique developed by the research team — Morcos said scientists can classify protein sequences by their evolutionary changes and their specific functions, then generate new sequences similar in composition, along with a rating of their compatibility with real-world function.
“Recent focus in the field has shifted toward using machine-learning approaches to predict protein structures and understand protein sequence attributes. The sequence space of proteins is astronomically large, so identifying viable sequences is a hard problem,” Morcos said.
Morcos and his team plotted protein-sequence data based on similar characteristics.
“The closer proteins are to each other in this virtual landscape, the more similar they are in function,” he said. “The map implies where we have a higher chance for a novel protein to be functional — there are many possible mutations as proteins evolve, but very few are fit to exist.”
The UTD researchers used mathematical methods to create peaks and valleys in the virtual landscape. These barriers represent sets of improbable sequences that help isolate groups of proteins in terms of their function or evolutionary trajectory, similar to how geographical boundaries can isolate groups of animals who then evolve differently from those in other isolated areas.
“Our new framework is like a road map. Rather than simply analyzing existing protein sequences, we look at the evolution of the proteins and construct maps looking both at proteins that already exist as well as generating and plotting out potential sequences.”
Dr. Faruck Morcos, associate professor of biological sciences in the School of Natural Sciences and Mathematics
Color-coding provides that third dimension of description of each coordinate. Proteins that already exist are also included and are concentrated in the dark areas.
“Is this protein fit to perform its function or not? How much does it look like a real protein? The dark blue regions are valleys of high fitness, where most proteins appear like things that can exist. These sequences might become real proteins,” Morcos said. “Brighter-colored regions are less explored and probably not very fit.”
Morcos said their system can also catalog proteins of unknown function in a process called annotation.
“The majority of protein sequences that exist don’t yet have an annotation — a label indicating a function or location,” he said. “We just don’t know what they do. That’s why scientists invest so much effort in accurately predicting the function of a protein. Our map is an effective way to infer the functions of a new protein by knowing what its neighbors do.”
Jonathan Martin MS’21, a molecular biology doctoral student, is co-first author of the study. A fourth UTD-affiliated author, postdoctoral scholar Dr. Claude Sinner, now works for Sitly in the Netherlands.
The research was supported by a grant (R35GM133631) from the National Institute of General Medical Sciences, a component of the National Institutes of Health, and by a Faculty Early Career Development Program (CAREER) grant (MCB-1943442) to Morcos from the National Science Foundation.
Media Contact: Stephen Fontenot, UT Dallas, 972-883-4405, stephen.fontenot@utdallas.edu, or the Office of Media Relations, UT Dallas, (972) 883-2155, newscenter@utdallas.edu.





